实操 | 合并VCF文件的几种方法及注意事项

 最新推荐文章于 2024-05-30 16:17:47 发布
最新推荐文章于 2024-05-30 16:17:47 发布
 阅读量9.7k
阅读量9.7k
 收藏
21
收藏
21


 点赞数
6
点赞数
6
背 景
在基因组分析领域的很多不同场景中,需要合并VCF文件。
VCF文件。简单来说,就是记录样本基因型的文件。但多数VCF文件不只记录了基因型,也包含有关该基因型的来源的细节。
其它文件。VCF文件的上游是BAM文件,主要记录Reads与参考基因组的比对信息;更上游的,就是FASTQ测序数据,以及物种的参考基因组。
不同类型的VCF文件。VCF文件有单样本的、多样本的;也有普通VCF文件 (只记录变异,未测到的、野生型的都不记录),及GVCF文件 (野生型的、变异的都记录,未测到的不记录)。
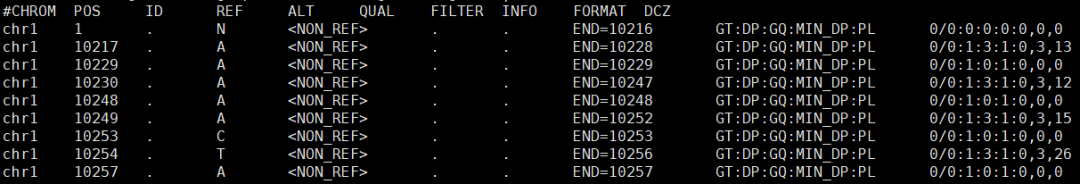
一个典型的GVCF文件
因此,本篇文章的使用者,需要首先了解GVCF文件与普通VCF文件的不同。因为二者对应的生信处理方法也非常不同。但具体有哪些不同,这里不再继续讲,可自行在网络资源上按照相应的关键词检索。
合并VCF文件需要注意的问题。VCF文件有时是多个样本、多个个体、或多个病例的合并;有时是不同染色体区域的VCF合并。上述每个场景都涉及不同的软件、程序,甚至算法,需要非常小心、谨慎地操作。
合并:不同样本的GVCF文件
GATK的CombineGVCFs + GenotypeGVCFs
上面2个程序是一套组合,不可拆分,不可单独使用。
那为什么GATK开发者将二者分开呢,推测有两个原因:① 二者分别有些特定的参数;② 第1个程序非常耗时,第2个程序相对较快、但算法复杂。这个问题对使用者也无关紧要。
# 合并队列中每个样本的每一个变异 (GVCF文件)
gatk CombineGVCFs \
-R $ref \
$(for i in `tail -n +2 metadata.txt | cut -f 1 `; do echo "--variant ${i}.hg38.raw.g.vcf " ;done) \
-O cohort.g.vcf.gz
# 获取具体基因型,完成变异Calling
gatk GenotypeGVCFs \
-R $ref \
--dbsnp ${dbSNP} \
--variant cohort.g.vcf.gz \
-O Genotype.cohort.dbSNP.g.vcf当样本很多、数据量也大时,CombineGVCF程序很消耗内存,并且一旦中断(文件不全)就得重新来。其"-L"参数 (如"-L chr1:1-10000") 也不推荐使用 (否则GenotypeGVCFs步骤可能报错),但"-L chr1"没问题。
解决方法:① 限制内存:用"--java-option Xmx20g"等;② 分染色体,用"-L chrX"等。③ 组合使用①和②。
需要了解的是,GenomicsDB可以替代:CombineGVCFs + GenotypeGVCFs,将多个样本GVCF处理生成一起的工作空间。两种方案各有各的优缺点。
根据GATK官网的描述,GenomicsDB更适用于几百个样本以上的情形。
合并:不同染色体区域的VCF文件
cat chr.list.25
# chr1
# chr2
# chr3
# ...
# chr22
# chrX
# chrY
# chrM
gatk MergeVcfs \
$(for i in `cat chr.list.25 | cut -f 1 `; do echo "-I Genotype.cohort.${i}.dbSNP.g.vcf " ;done) \
-O Genotype.cohort.dbSNP.g.vcfMergeVcfs与CombineGVCFs不同。前者用于单纯地合并:样本相同、位点独立的VCF文件。如:同一个(或一组)样本的不同染色体的结果。
不像CombineGVCFs,MergeVcfs不做"gVCF block"的计算。此外MergeVcfs会检测两个VCF文件里的样本名是否相互"match"。
如果只查看MergeVcfs程序的介绍,根本看不出来它的用法的特点 (例如:对GVCF文件的合并可能无效),那么必然容易踩坑:
MergeVcfs (Picard) - Combines multiple variant files into a single variant file.
事实上,MergeVcfs及其等价的程序 (GatherVcfs) 不可用于合并不同样本的GVCF文件。但用来合并不同基因组区域的文件非常方便。
此场景除了MergeVcfs、GatherVcfs外的其它程序:
vcf-concat sample1.chr1.vcf sample.chr2.vcf ... > sample1.chrAll.vcf
bcftools concat sample1.chr1.vcf sample.chr2.vcf ... -o sample1.chrAll.vcf其中,通过conda安装的vcftools,可能不带vcf-concat等程序。从这一点看,bcftools更方便。
1个经验是:既然有GATK的MergeVcfs可用,那就尽量不用vcftools或bcftools,否则可能踩到另一个坑:不同程序对VCF文件的索引格式的要求不同、VCF的"FORMAT"列等也可能改变。
合并:不同样本的普通VCF文件
普通VCF文件只记录变异,即:① 无0/0基因型 (测序测到了、但未变异,即"Wild type") ;② 无"./."基因型 (即"缺失",测序未测到,即"No call") 。
对不同样本的⽂件合并,共有位点会合并、统计;非共有位点若在某1个样本中无变异,则会⾃动记为缺失 ("./.") 。
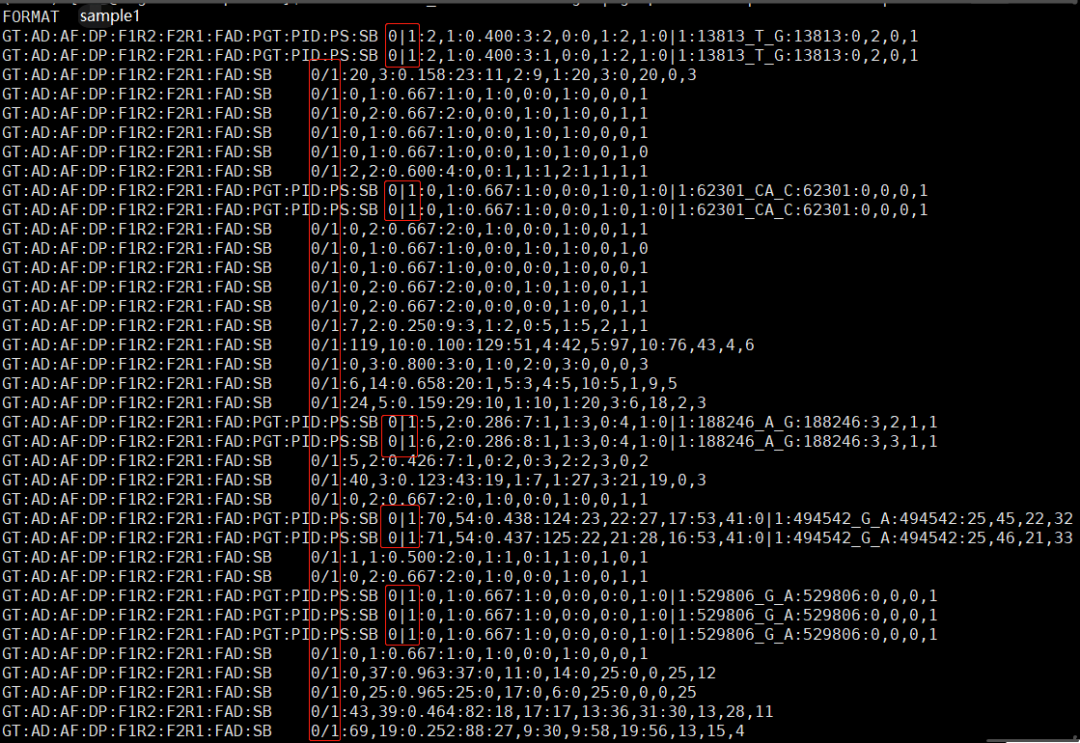
1个典型的普通VCF文件 (只查看了第9、10列)
vcftools和bcftools在使用之前一般都需要对VCF文件:压缩、索引 (略)。
vcf-merge # 略 (对于连软件安装都麻烦的程序)
bcftools sample1.vcf sample2.vcf ... -o sample.all.vcf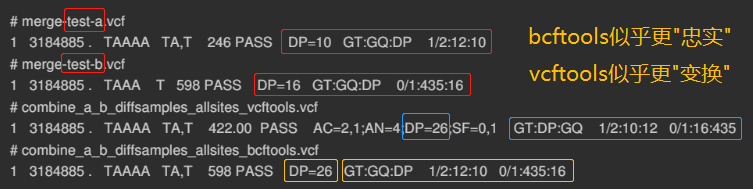
vcf-merge重新计算了AC、AN等指标的值
合并分型质量 ("QUAL"列) 时,vcf-merge取了平均取值,bcftools取了最⼤值, (下图的)gatk CombineVariants (不是CombineGVCFs,也不是MergeVcfs/GatherVcfs)取了最⼩值 (gatk4)
图片来源:https://wenku.baidu.com/view/a0ecad5602f69e3143323968011ca300a7c3f643.html

gatk CombineVariants (GATK4已无此程序)
# 压缩、索引单个VCF文件
ls sorted.*.vcf | while read id;do
bcftools view $id -Oz -o $id.gz
bcftools index $id.gz
done
# 合并
bcftools merge --threads 8 -m id -O z sorted.*.vcf.gz \
-o bcftools.merged.103samps.vcf.gz
# -m, --merge (关于多等位基因)Allow multiallelic records for <snps|indels|both|all|none|id>, see man page for details [both]
# -O, --output-type <b|u|z|v> 'b' compressed BCF; 'u' uncompressed BCF; 'z' compressed VCF; 'v' uncompressed VCF [v]
# gVCF参数(可能不适用于gVCF文件):
# -g, --gvcf <-|ref.fa> merge gVCF blocks, INFO/END tag is expected. Implies -i QS:sum,MinDP:min,I16:sum,IDV:max,IMF:max
# 测试某个位点
# zcat bcftools.merged.103samps.vcf.gz | grep '1 12626 '
# 有返回结果。但无DP等信息
# 索引合并后的文件
bcftools index bcftools.merged.103samps.vcf.gz &bcftools merge虽然有"--gvcf"参数,但根据之前的测试,可能不适用于对gVCF文件的合并。
总 结
总之,① 合并VCF文件要区分其文件类型,如:是否为gVCF文件,是否为基因组的不同区域,其内部的样本名称等;② 考虑到整个流程的兼容性和流畅性,建议当GATK有相应的工具时,优先使用之;③ 其它场景可依次考虑:bcftools、vcftools。
往期精品(点击图片直达文字对应教程)
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |




























机器学习
后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集
















 1万+
1万+































 到【灌水乐园】发言
到【灌水乐园】发言










普通网友: 干货满满,实用性强,博主的写作风格简洁明了,让人一目了然。文章涵盖了很多实用的知识点。【我也写了一些相关领域的文章,希望能够得到博主的指导,共同进步!】
多巴胺81: 网络 请求otu_table.txt源文件格式
2301_78491303: 老师您好,请问这个signac安装包如何下载呢?并且相关的数据包能不能导入到Rstudio里面进行分析啊?
MR. Paw: 月薪才六千新币起?
是海绵宝宝吖: 博主好,dcast功能图示的图没有显示欸